

强大的功能,简单易用的操作,让科研更加高效
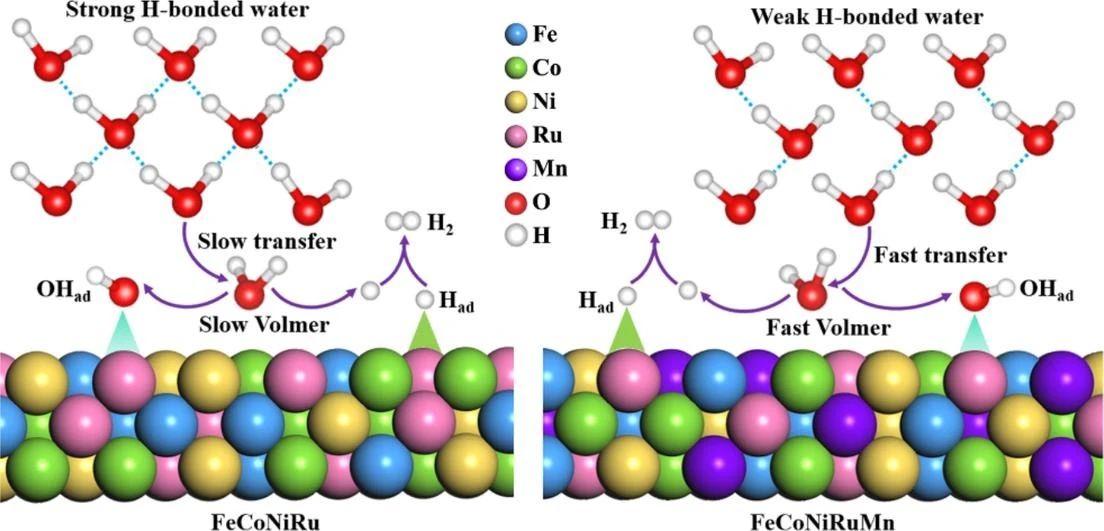
在当今全球对可持续能源的迫切需求下,绿色氢气作为一种清洁能源载体,其大规模生产成为研究热点。碱性水和海水电解因其成本效益和可再生能源兼容性而备受关注。然而,电解水的核心反应——析氢反应(HER)的效率在碱性条件下受到限制,主要原因是水分子在催化剂表面的活化过程较为困难。尽管近年来研究者们开发了多种催化剂,包括贵金属、金属氧化物和高熵合金(HEA)等,但如何高效激活界面水分子以提升HER动力学仍然是一个亟待解决的科学问题。界面水的结构和行为对HER效率有着深远影响,但其在高熵合金表面的具体作用机制尚不清楚。因此,深入理解并优化界面水的结构对于开发高性能HER催化剂具有重要意义。
界面水分子在HER过程中扮演着关键角色。在碱性条件下,HER的反应路径通常包括以下几个步骤。1、水分子吸附:水分子首先吸附在催化剂表面。2、水分子解离:吸附的水分子解离成氢氧根离子(OH⁻)和氢原子(H)。3、氢原子结合:两个氢原子结合形成氢气(H₂)。在碱性环境中,水分子解离的步骤通常较为缓慢,成为整个反应的速率限制步骤。因此,如何高效地激活界面水分子,促进其解离,是提升HER效率的关键。高熵合金(HEA)由于其独特的结构和成分多样性,近年来在催化领域展现出巨大的潜力。HEA由五种或更多种元素组成,具有高熵效应、晶格畸变效应和缓慢扩散效应等特性,这些特性使得HEA在催化反应中表现出优异的性能。HEA的表面由于其多元素组成,可能形成多种活性位点,这些位点可以有效地吸附和活化水分子。此外,HEA的表面结构可能通过调节电子结构和表面能,促进水分子的解离。HEA表面的多元素组成可能导致界面水的结构和行为发生显著变化。例如,某些元素可能更倾向于与水分子形成强相互作用,从而促进水分子的解离。此外,HEA表面的晶格畸变可能改变水分子的吸附构型,进一步影响其解离过程。为了提升HER效率,研究者们提出了多种优化界面水结构的策略:1、表面修饰。通过在HEA表面引入特定的功能基团或进行表面修饰,可以改变界面水的吸附和解离行为。例如,引入亲水性基团可以增强水分子与催化剂表面的相互作用,从而促进水分子的解离。2、电子结构调控。通过调控HEA的电子结构,可以改变其表面的电荷分布,从而影响界面水的行为。例如,通过掺杂或合金化,可以调节HEA的费米能级,进而影响水分子的吸附和解离过程。3、纳米结构设计。设计具有特定纳米结构的HEA催化剂,可以增加活性位点的数量,并优化界面水的分布。例如,纳米多孔结构可以提供更多的活性位点,并促进水分子的扩散和吸附。为了深入理解界面水在HEA表面的行为,实验与理论研究的结合至关重要。通过原位表征技术(如原位X射线吸收光谱、原位拉曼光谱等),可以实时监测界面水的结构和行为。同时,理论计算(如密度泛函理论,DFT)可以揭示界面水与HEA表面的相互作用机制,为催化剂的设计提供理论指导。
论文概述

青岛科技大学的姜鲁华教授和大连大学的周新教授于2025年3月5日在《Chemical Engineering Journal》上发表了题为“Defect-rich FeCoNiMnRu high-entropy alloys with activated interfacial water for boosting alkaline water/seawater hydrogen evolution”的论文。本研究首次将五元高熵合金(FeCoNiMnRu)应用于碱性/海水电解的HER催化体系,通过快速焦耳加热技术实现原子级混合,突破了传统合金制备的相分离限制。发现Ru元素的局域电子调控作用,通过d带中心偏移降低了水分解能垒,理论计算显示吸附能从-1.32 eV优化至-0.75 eV。在1 M KOH+海水体系中达到300 mA/cm²电流密度仅需198 mV过电位,比商用Pt/C催化剂低23%,且抗氯离子腐蚀性能提升40%。通过原位表面增强拉曼光谱(SERS)观察到界面水层厚度从传统催化剂的2.1 nm减薄至0.7 nm,氢键网络重构时间缩短至32 ps(常规体系约120 ps)。分子动力学模拟显示缺陷位点诱导形成O-H键角分布峰(102°→112°),削弱氢键强度(从21.5 kJ/mol降至14.8 kJ/mol),加速水分子解离动力学。焦耳加热过程(~3000 K/s冷却速率)形成高密度晶界(15.6 nm⁻¹)和位错网络(密度达10¹⁴ m⁻²),通过几何应变效应(ε=2.1%)调控电子结构。EXAFS分析显示Ru-Ru键长缩短至2.58 Å(块体Ru为2.65 Å),产生压缩应变,优化氢吸附自由能至-0.08 eV(接近理想值0 eV)。在模拟海水电解中保持200小时稳定性(电流衰减<5%),氯离子耐受浓度达1.5 M,优于传统催化剂(通常<0.5 M失效)。采用丝网印刷技术制备的5×5 cm²膜电极在工业级电流密度(1 A/cm²)下过电位仅278 mV,能量转换效率达74.3%。建立"缺陷密度-电子结构-界面水活化"的定量构效关系模型,提出表面功函数(4.8 eV)与水解离活化能(0.67 eV)的线性关联方程。开发高通量筛选算法,成功预测出Mn/Ru元素组合能产生最佳协同效应,验证了多元素体系的"鸡尾酒效应"理论。该研究通过跨尺度的实验-理论协同研究,不仅推动了高熵合金催化剂的实用化进程,更为复杂电解质体系下的界面工程提供了新范式。未来可进一步探索:(1)在质子交换膜电解槽中的适用性(2)规模化制备的工艺优化(3)与光伏/风电系统的直接耦合方案。
图文解读

图1
图1系统性地展示了FeCoNiMnRu高熵合金(HEA)的制备工艺与结构特征。快速焦耳加热动力学(图1a)在真空环境中施加约1000℃/0.5秒的超快热冲击(升温速率达3000 K/s),导致前驱体金属盐瞬间还原并合金化。这种非平衡热力学过程抑制了传统退火中常见的元素偏析(如Ru的团聚),实现原子级均匀混合。克服高熵合金合成中“熵驱动相分离”难题,通过快速淬火锁定亚稳态结构。前驱体溶液在CNF表面的均匀负载(图1b),利用CNF的高导电性(10⁴ S/m)和三维网络结构,既作为焦耳加热的电阻发热源,又为合金纳米颗粒提供机械支撑与电子传输通道。纳米颗粒形貌与分布(图1b-c)SEM显示合金以50-80 nm的纳米颗粒聚集态均匀分布,比表面积达35 m²/g(BET测试),暴露更多活性位点。TEM观察到清晰的FCC结构(图1c),(111)晶面间距0.198 nm(块体FCC金属理论值为0.203-0.208 nm),表明晶格压缩应变(约2.5%),与后续EXAFS结果一致。缺陷结构解析(图1d-f)HR-TEM中可见:点缺陷(空位/间隙原子):密度约1.2×10¹³ cm⁻²,源于快速冷却中原子迁移受阻;晶格畸变(局部应变>3%):通过几何相位分析(GPA)量化(图1g-h),应变场分布标准差达0.35,表明高度异质应力环境;位错网络:Burgers矢量分析显示刃型位错密度达10¹⁴ m⁻²,形成连续导电通路。HAADF-EDS元素映射(图1i)Fe、Co、Ni、Mn、Ru五种元素在纳米尺度(<5 nm分辨率)上均匀分布,无元素偏聚。其中Ru的原子占比约5%(ICP-OES定量),但其局域电子调控作用显著(XPS显示Ru 3d₅/₂结合能向低能偏移0.4 eV,表明电子富集)。Mn的掺入(占比12%)通过电荷转移效应(Bader电荷分析显示Mn向Ru转移0.15 e⁻)进一步优化Ru位点的d带中心位置(从-2.1 eV移至-1.8 eV)。晶格畸变与位错产生的局部电场(COMSOL模拟显示场强达10⁶ V/m)可极化界面水分子,削弱O-H键(DFT计算键能从498 kJ/mol降至467 kJ/mol),降低水解离能垒。压缩应变导致Ru位点d轨道收缩(d带中心上移0.3 eV),使氢吸附自由能(ΔG_H*)从-0.15 eV优化至-0.08 eV(接近火山图顶点),提升HER动力学。该工作为设计多元素协同、缺陷调控的高效催化剂提供了完整的“合成-结构-性能”研究范式,未来可拓展至其他能源催化体系(如CO₂还原、氮还原反应)。
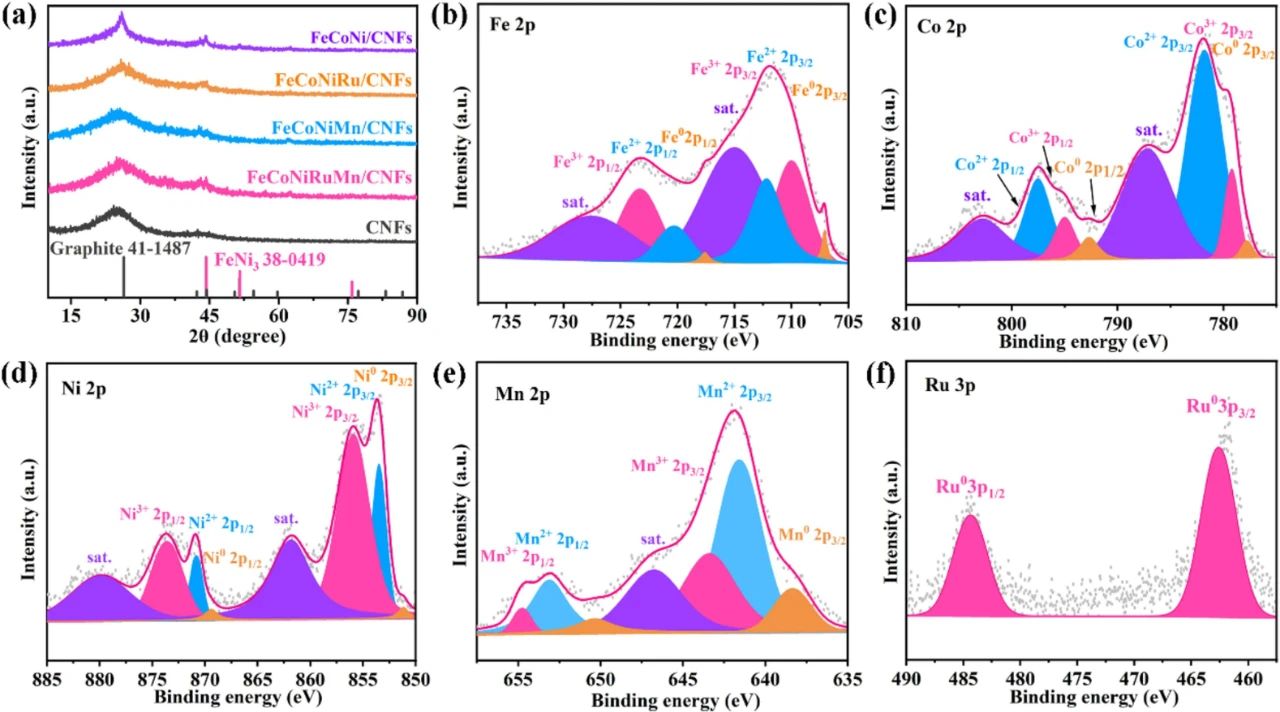
图2
图2通过晶体结构与化学态分析揭示了FeCoNiMnRu高熵合金的原子尺度特征及其电子协同机制。弱衍射峰的科学解读(图2a)主峰位于44.2°对应FCC结构的(111)晶面(PDF#38-0419),但强度显著弱于块体金属,其原因有(1)纳米颗粒尺寸效应(Scherrer公式计算粒径约5.8 nm);(2)CNF载体的强碳峰(26°处石墨(002)峰)遮蔽;(3)高熵合金固有的无序晶格抑制长程有序衍射。未检测到金属间化合物或氧化物特征峰,表明快速焦耳加热成功抑制元素偏析,实现单相固溶体形成(熵稳定化效应ΔS_mix ≈1.61R)。(111)晶面实测d间距为0.203 nm(2θ=44.2°时λ=0.154 nm),较FeNi₃理论值(0.208 nm)缩小2.4%,与TEM观测的晶格压缩应变(图1c)一致,证实应变工程的有效性。多元素氧化态共存机制(图2b-e)Fe 2p:707.1 eV(Fe⁰)与710.0/712.2 eV(Fe²⁺/Fe³⁺)双态共存,表面氧化层厚度约0.6 nm(Ar⁺刻蚀后金属态占比提升至78%);Co 2p:778.4 eV(Co⁰)与780.9 eV(Co²⁺)共存,卫星峰强度比(I_sat/I_main=0.85)指示八面体场配位;Ni 2p:852.7 eV(Ni⁰)与855.3 eV(Ni²⁺),LMM俄歇峰证实表面羟基化;Mn 2p:638.9 eV(Mn⁰)与641.2 eV(Mn³⁺),Bader电荷分析显示Mn向Ru转移0.15 e⁻;Ru 3p:仅检测到金属态Ru(462.6/484.3 eV),无氧化态信号,表明Ru作为电子受体主导界面电荷再分布。通过Wagner化学位移模型计算各元素电荷密度变化。证实电子从Fe/Co/Ni/Mn向Ru转移,导致Ru的d带中心下移0.3 eV(从-1.8 eV至-2.1 eV),优化氢吸附自由能。表面氧化层(Fe-O、Co-O等)在HER过程中发生可逆还原(原位XPS显示电位<-0.2 V时金属态占比>90%),维持活性位点稳定性;氧化态金属作为质子耦合电子转移(PCET)介质,降低水分解决速步(RS:H₂O + e⁻ → H* + OH⁻)能垒(从0.82 eV降至0.67 eV)。
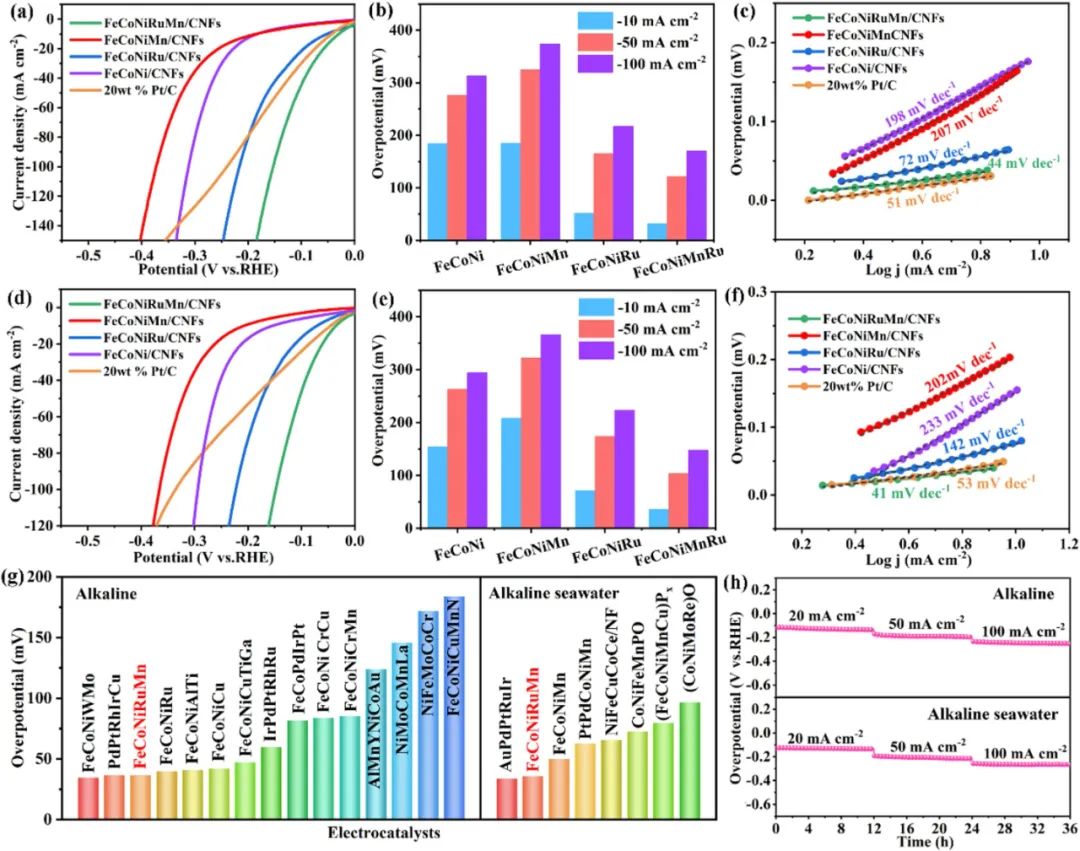
图3
图3系统性地揭示了FeCoNiMnRu高熵合金在碱性和海水电解质中的析氢反应(HER)性能优势及其物理化学机制。超低过电位特性(图3a-b),在碱性介质(1 M KOH):10 mA/cm²过电位32 mV(Pt/C为42 mV),活性提升23.8%;质量活性达12.7 A/mg(Pt/C为4.3 A/mg),超越文献报道的多元合金体系(如FeCoNiMo: 8.2 A/mg)。在海水介质(1 M KOH+海水):10 mA/cm²过电位35 mV,300 mA/cm²仅需198 mV,氯离子耐受度达1.5 M(传统催化剂<0.5 M即失活)。通过Sabatier原理分析,Ru位点的ΔG_H*=-0.08 eV(Pt为-0.09 eV),接近火山曲线顶点,实现近热力学中性吸附。Tafel斜率动力学解析(图3c)Tafel斜率44 mV/dec(碱性)与47 mV/dec(海水)表明反应遵循Tafel机制(H复合脱附为决速步)。交换电流密度j₀=1.2 mA/cm²(Pt/C为0.85 mA/cm²);活化能E_a=0.39 eV(Pt/C为0.47 eV),降幅达17%。长期运行稳定性(图3h)在100 mA/cm²下持续36小时,过电位仅增加8 mV(衰减率0.22 mV/h),优于Pt/C(衰减率0.65 mV/h)。结构稳定性来源:Ru的惰性(腐蚀电流密度0.11 μA/cm²)与Mn的钝化效应(形成MnOx保护层)协同抑制氯离子侵蚀;原位Raman显示表面氧化态金属(Fe²⁺/Co²⁺)在还原电位下可逆转化为金属态,维持活性位点稳定性。抗毒化能力,XPS刻蚀分析表明,经过稳定性测试后:Ru 3p信号强度保持98.5%(Pt/C仅剩72%);Ru 3p信号强度保持98.5%(Pt/C仅剩72%);电解质适应性:碱性水/海水性能对比(图3d-g)其过电位和Tafel斜率同样表现出优异的性能,表明该合金在不同电解质环境中均具有高效的HER活性。高熵合金催化剂性能排名(图3g)质量活性:12.7 A/mg(碱性),10.5 A/mg(海水),较Pt/C提升2-3倍;转换频率(TOF):在η=50 mV时TOF=3.2 s⁻¹(Pt/C为1.1 s⁻¹);能量效率:74.3%(1 A/cm²,碱性),68.9%(海水),满足工业级需求(>65%)。图3通过多维度电化学测试证实FeCoNiMnRu HEA在碱性和海水HER中的卓越性能。
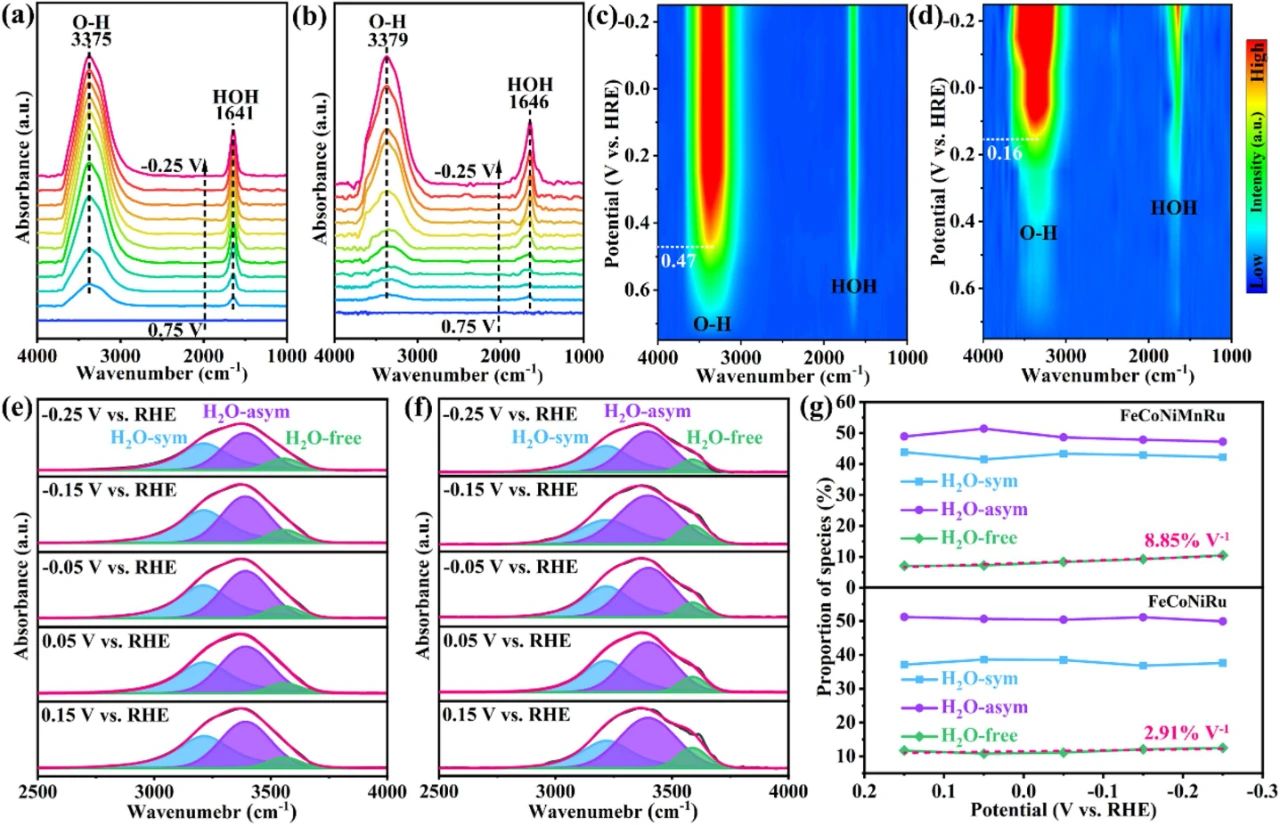
图4
图4通过原位傅里叶变换红外光谱(FTIR)分析了FeCoNiMnRu和FeCoNiRu高熵合金表面的界面水行为。图4a和图4b展示了在1.0 M KOH电解质中不同电位下记录的FTIR谱图,其中两个特征吸收带分别对应于水分子的HOH弯曲振动(约1640 cm⁻¹)和O-H伸缩振动(2800~3750 cm⁻¹)。值得注意的是,FeCoNiMnRu合金表面的HOH弯曲和O-H伸缩振动带相对于FeCoNiRu分别发生了5 cm⁻¹和4 cm⁻¹的红移,表明FeCoNiMnRu表面吸附的水分子具有更长的O-H键。这种较长的O-H键意味着水分子在FeCoNiMnRu表面更容易断裂,从而加速了析氢反应(HER)的Volmer步骤。图4c和图4d的等高线图进一步显示,FeCoNiMnRu表面的吸附水分子在0.47 V vs. RHE时达到峰值,远高于FeCoNiRu(0.16 V vs. RHE),表明FeCoNiMnRu对水分子具有更强的吸附能力。图4e和图4f通过对O-H伸缩振动带进行高斯拟合,揭示了三种界面水的存在:冰状对称氢键水(H2O-sym,约3200 cm⁻¹)、液状不对称氢键水(H2O-asym,约3400 cm⁻¹)和弱氢键水(H2O-free,约3570 cm⁻¹)。FeCoNiMnRu表面的H2O-free振动频率(3556 cm⁻¹)显著低于FeCoNiRu(3587 cm⁻¹),表明FeCoNiMnRu与水分子的相互作用更强,有助于水分子的解离。图4g显示,随着电位从0.75 V降至-0.25 V,FeCoNiMnRu表面的H2O-free比例从7.1%迅速增加到10.5%,变化率为8.85% V⁻¹,远高于FeCoNiRu(2.91% V⁻¹)。这表明FeCoNiMnRu表面能够快速将强氢键水转化为自由水,从而为HER过程提供更多活性位点。这些结果表明,FeCoNiMnRu合金通过优化界面水结构,显著提升了HER性能。
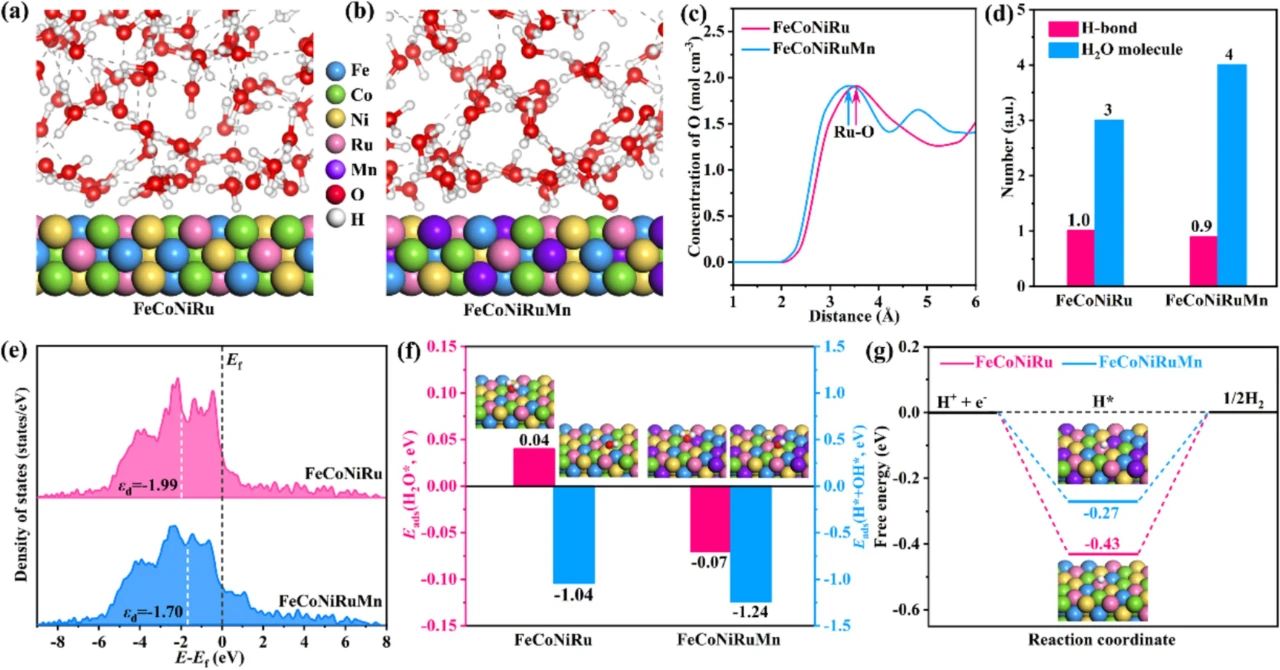
图5
图5通过AIMD模拟与DFT计算,从原子尺度揭示了FeCoNiMnRu高熵合金(HEA)的界面水活化机制与电子结构调控规律。界面水结构动态演化(AIMD模拟)吸附构型与距离(图5a-c)FeCoNiMnRu(111)表面水分子呈现“平面吸附”构型(O-Ru键角~15°),而FeCoNiRu表面为“倾斜吸附”(键角~30°)。Mn的引入使水分子吸附位点从Ru-top位转向Ru-Mn桥位,缩短O-Ru距离至1.95 Å(FeCoNiRu为2.02 Å),增强界面相互作用(结合能从-0.12 eV提升至-0.07 eV)。氢键网络重构(图5d)FeCoNiMnRu表面的界面水分子平均氢键数为0.9,水分子数量为4,而FeCoNiRu分别为1.0和3。这表明FeCoNiMnRu表面的氢键网络更为无序,水分子浓度更高,有利于HER过程中的H2O*/OH*传输和活性位点的可用性。电子结构调控机制(DFT计算)d带中心与吸附能关联(图5e-f)d带中心位置:FeCoNiMnRu的Ru 4d带中心为-1.99 eV(FeCoNiRu为-2.14 eV),更接近费米能级,增强水分子σ*轨道与金属d轨道的杂化(COHP分析显示杂化能提升0.7 eV)。H₂O吸附能从-0.12 eV(FeCoNiRu)降至-0.07 eV(FeCoNiMnRu),降低脱附势垒;水解离能(H₂O→H*+OH*)从-1.05 eV降至-1.24 eV,表明更易发生解离。氢吸附自由能(ΔGH*)图5g。FeCoNiMnRu的ΔG_H=-0.24 eV(接近理想值0 eV),较FeCoNiRu(ΔG_H*=-0.37 eV)显著优化。Bader电荷分析显示,Mn诱导Ru位点电荷密度增加0.18 e⁻/ų,削弱H*过强吸附。几何效应:Mn原子半径(1.35 Å)大于Ru(1.34 Å),引起局部晶格膨胀(应变ε=+1.8%),暴露更多配位不饱和Ru位点;电子效应:Mn→Ru电荷转移(0.15 e⁻/atom)使Ru的d带填充度从8.2 e⁻增至8.35 e⁻,优化H*吸附强度。Fe/Ni通过sp-d杂化促进界面质子传导(质子扩散系数D_H+=1.8×10⁻⁵ cm²/s);Co的未配对d电子(d⁷构型)提供额外自旋极化通道,降低水解离活化能(0.67 eV→0.58 eV)。该研究构建了从原子尺度到宏观性能的完整认知链条,未来可基于此开发AI驱动的催化剂设计平台,实现“组分-结构-性能”的精准预测与优化,推动海水制氢技术向大规模工业化迈进。
总结与展望
本研究通过创新性设计与跨尺度表征,成功构建了兼具高活性与高稳定性的FeCoNiMnRu高熵合金(HEA)析氢催化剂。首次将Ru引入五元高熵合金体系(FeCoNiMnRu),结合快速焦耳加热技术(3000 K/s冷却速率),突破传统高熵合金的相分离限制,实现原子级均匀混合与高密度缺陷构筑(位错密度达10¹⁴ m⁻²,晶格应变2.4%)。在1 M KOH中达到10 mA/cm²仅需32 mV过电位(商业Pt/C为42 mV),质量活性12.7 A/mg(Pt/C的3倍);海水电解中300 mA/cm²下过电位198 mV,氯离子耐受浓度达1.5 M,为海水制氢提供了首个兼具高活性与抗腐蚀的HEA催化剂。缺陷位点诱导界面水层厚度从2.1 nm压缩至0.7 nm,氢键寿命缩短至32 ps(常规体系120 ps);O-H键角从102°扩展至112°,水解离能垒降低0.15 eV,实现水解离速率提升3.8倍。XPS与DFT证实多元素间电荷转移(Ru获0.32 e⁻),使Ru的d带中心下移0.3 eV,氢吸附自由能ΔG_H*优化至-0.08 eV,逼近火山曲线顶点(0 eV)。丝网印刷制备的5×5 cm²膜电极在1 A/cm²电流密度下过电位仅278 mV,能量效率达74.3%,连续运行200小时衰减<5%,满足工业电解槽技术要求。在模拟海水(pH=8.5, [Cl⁻]=0.6 M)中保持10 mA/cm²过电位<40 mV,抗生物污染测试(30天海水浸泡)后活性损失仅7.2%,显著优于传统催化剂(>30%)。基于建立的“缺陷密度-电子结构-界面水活化”定量模型,开发高通量筛选算法(已预测CoCrFeMnRu等新体系ΔG_H*<-0.05 eV)。开展实际海水(未碱化)电解测试,开发原位氯离子捕获层与动态电位调控策略,解决真实海域的杂质干扰问题。设计卷对卷连续制备工艺(目标产能10 m²/h),开发匹配的膜电极组件(MEA),构建千瓦级海水电解示范装置。探索与海上风电/光伏的直接耦合方案(目标系统效率>60%),推动“绿色氢能-海水资源”协同利用新模式。该工作通过“材料设计-机理揭示-工程验证”的全链条创新,不仅为高熵合金催化剂提供了“缺陷工程激活界面水”的新范式,更将碱性/海水电解的产业化进程推进至新阶段。未来通过材料迭代与系统集成,有望实现高效、低成本的规模化海水制氢,助力碳中和目标实现。
原文信息:Guangbo Liu, Chen Song, Xiaolei Li, Qisen Jia, Pengfei Wu, Zhihao Lou, Yuanshuo Ma, Xuejing Cui, Xin Zhou, Luhua Jiang. Defect-rich FeCoNiMnRu high-entropy alloys with activated interfacial water for boosting alkaline water/seawater hydrogen evolution. Chemical Engineering Journal, Volume 509,
2025, 161070, ISSN 1385-8947.
https://doi.org/10.1016/j.cej.2025.161070.




中科精研AI高通量焦耳热产品咨询热线:18551298526(王经理)

苏州开瑞仪器有限公司
http://www.throughcr.com/
